



高熵氧化物(HEOs)是由多种金属阳离子形成的固溶体,具高熵效应、晶格畸变和多元素协同作用。而DFT作为核心工具,通过GGA/PBE、HSE06等泛函,研究其晶格优化、电子结构等。不同晶体结构HEOs计算焦点不同,如岩盐型的晶格畸变,萤石型的相稳定性等,其在OER催化等领域应用前景广阔。
什么是高熵氧化物?
高熵氧化物是指由多种金属阳离子在单一晶体结构中形成的固溶体,其构型熵需满足Sconf≥1.5R(R为气体常数),当所有阳离子等摩尔混合时最大熵可达1.61R。
这类材料的独特性质由三大核心效应驱动:高熵效应通过熵增主导相稳定性,有效抑制元素偏析;晶格畸变源于阳离子尺寸差异形成的局部应力场,进而影响电子结构;多元素协同作用导致离子迁移率降低,呈现缓慢扩散特性,元素间的协同作用形成“鸡尾酒效应”,使其性能超越单一组分材料。
高熵氧化物的结构多样性是显著优势,例如岩盐结构(R-HEO)的(Co,Cu,Mg,Ni,Zn)O,其研究重点包括晶格畸变能与形成焓;萤石结构(F-HEO)的(Ce,Zr,Hf,Pr,La)O₂,关注氧空位形成能与离子迁移路径。
钙钛矿结构(PE-HEO)的(La,Pr,Nd,Sm,Gd)CrO₃,侧重能带调控与铁电性;尖晶石结构(SP-HEO)的(Cr,Mn,Fe,Co,Ni)₃O₄,则聚焦磁序与电子自旋态等。
这些结构类型通过密度泛函理论 (DFT) 计算可深入探究其物理化学机制,为功能材料设计提供理论支撑。

DOI:10.1021/acsenergylett.4c01129
DFT在HEOs中的应用
在高熵氧化物(HEOs)的研究中,密度泛函理论(DFT)是揭示其微观机制与性能调控的核心计算工具,其应用涵盖计算方法选择、泛函优化及多维度物理化学性质的理论预测。
在计算方法与泛函选择方面,主流泛函类型各有侧重:GGA/PBE泛函因计算效率高且晶格常数预测误差小于2%成为基础方法,但存在带隙低估问题;HSE06泛函通过混合泛函修正带隙偏差,适用于析氧反应(OER)催化剂活性预测等需精确带隙的场景,然其计算成本较高;SCAN/r²SCAN泛函则在强关联体系描述上更优,尤其适用于过渡金属HEOs中电子强关联效应的处理,精度优于传统PBE泛函。
针对局域d/f电子主导的体系,如CoO₆八面体畸变问题,DFT+U方法通过引入Hubbard U参数修正局域关联能;而DFT+DMFT(动态平均场理论)则可捕捉NiO基HEOs中金属-绝缘体转变等动态关联效应。
在计算目标与典型结果层面,晶格优化旨在通过DFT弛豫预测HEOs稳定相结构,例如对比PBE优化得到的晶格常数与实验值,验证结构稳定性。
电子结构分析包括能带结构与态密度(DOS)计算,能带结构可确定带隙类型(直接/间接)及载流子有效质量,如通过GGA/SCAN/QSGW方法对比Cd₃As₂的能带结构以显示带隙修正效果,态密度分析则能揭示轨道贡献与费米能级位置,如NiO/MnO体系的总DOS及轨道分波DOS可明确O-p与过渡金属d轨道的杂化程度。
缺陷形成能计算结合热力学模型,用于评估氧空位浓度与离子电导率,为空位迁移机制提供理论依据;表面吸附能计算则通过机器学习辅助,识别催化活性位点,例如分析H₂在岩盐HEO (100) 晶面的吸附能分布,为OER/HER催化剂设计提供指导。
这些计算通过图表可视化,如晶格常数对比图、能带结构叠合图、态密度分波图及缺陷形成能流程图等,形成从原子结构到宏观性能的理论关联,为HEOs的成分设计、结构调控及功能优化提供量化支撑。

DOI:10.33774/chemrxiv-2021-gwm9m
HEOs体系分类与计算焦点
在高熵氧化物(HEOs)体系中,不同晶体结构类型的DFT计算焦点与其物理化学特性紧密关联。
岩盐型HEOs(R-HEOs)以(Co₀.₂Cu₀.₂Mg₀.₂Ni₀.₂Zn₀.₂)O为典型体系,采用PBE泛函优化得到的晶格常数约为4.27 Å,与实验值误差小于1%,通过键长分布标准差大于0.1 Å的分析,证实了阳离子尺寸差异导致的局部晶格畸变效应,该畸变形成的应力场对电子结构调控具有关键作用。
萤石型HEOs(F-HEOs)如 (Ti,Zr,Hf,Sn)O₂体系,DFT计算揭示其α-PbO₂结构的焓值低于金红石相,表明熵效应主导相稳定性选择,而阳离子尺寸失配引发的晶格应变能超过50 meV/atom,这种应变能通过影响氧空位形成与迁移路径,进而调控离子传导性能。
钙钛矿型HEOs(PE-HEOs)以La(Co,Cr,Fe,Mn,Ni)O₃为代表,其DFT研究聚焦于能带工程与氧空位迁移机制:通过Fe/Mn 掺杂调控O-p带中心位置,可优化析氧反应(OER)催化活性;采用NEB(nudged elastic band)方法计算得到氧空位迁移势垒低于0.8 eV,揭示了该体系在离子输运方面的优势。
这些基于DFT的计算研究,从晶格畸变、相稳定性、能带调控及缺陷迁移等维度,建立了HEOs组成-结构-性能的理论关联,为多组元氧化物材料的设计与优化提供了定量理论支撑。

DOI:10.1038/s43246-023-00372-5
OER催化剂的应用
在高熵氧化物(HEOs)作为氧析出反应(OER)催化剂的研究中,通过密度泛函理论(DFT)对尖晶石型 (Co,Ni,Fe,Mn,Cr)₃O₄的催化活性进行理论筛选,可有效规避传统实验试错的高成本。
该研究首先构建尖晶石 (111) 表面的4×4超胞模型(含192个原子),随机分配阳离子位点以满足等摩尔比组成;采用VASP软件结合PBE+U泛函(其中U_{Co}=3.5 eV、U_{Ni}=6.0 eV)进行计算,设置520 eV截断能与4×4×1的K点网格参数。
关键计算包括吸附能与反应自由能:吸附能Eads通过表面吸附前后能量差及O₂分子能量校正(补偿PBE对O₂结合能的低估)获得,反应自由能则借助计算氢电极(CHE)模型分析*OH→*O→OOH的各步ΔG。
结果可视化呈现OER自由能阶梯图与O吸附能分布直方图:前者显示Mn位点主导的O→OOH步骤为速率决定步骤(ΔG=1.8 eV),后者以多峰分布(Eads 范围−1.5 至−0.5 eV)证实表面活性位点的非均一性。
DFT计算揭示了Mn-Co协同作用可降低OOH形成能,为高活性HEO催化剂的组分设计与性能优化提供了量化理论依据,凸显了理论计算在指导多组元功能材料开发中的关键价值。
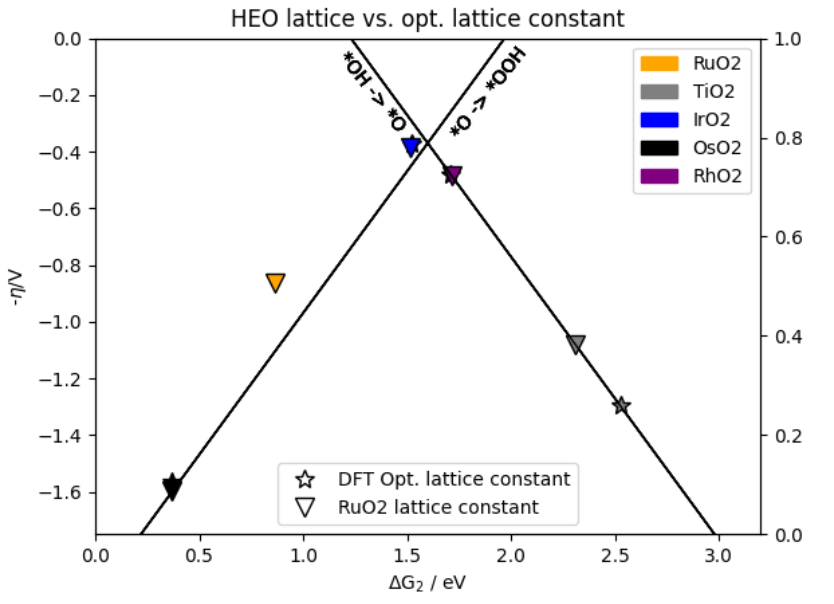
DOI:10.26434/chemrxiv-2022-7wwnh
总结
在高熵氧化物(HEOs)研究中,描述符工程通过14个几何与电子描述符(如d 带中心、配位数等)构建预测模型,实现H₂吸附能的高精度预测,平均绝对误差(MAE)低至0.06 eV。
此外,结合Materials Project数据库开展的高通量筛选,已成功构建HEOs形成能与带隙的预测模型。密度泛函理论(DFT)作为解析HEOs构效关系的核心工具,正从晶格稳定性分析向催化机制阐释等原子尺度研究深入。
未来研究需聚焦更精确的强关联计算方法(如SCAN+DMFT)开发,并深化机器学习辅助的高通量设计策略,以此推动HEOs在能源存储与转化领域的应用突破,实现从理论预测到实际应用的关键跨越。
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。

官方微信
《腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606
- 邮箱:fsfhzy666@163.com
- 腐蚀与防护网官方QQ群:140808414

“海洋金属”——钛合金在舰船的

腐蚀与“海上丝绸之路”