



共价有机框架,实现全波段太阳能转化
传统光催化技术长期受限于单重态(S₁)或三重态(T₁)激发态的选择性利用,导致能量转化效率与反应范围受限。尽管S₁态在电子转移驱动氧化还原反应中具有优势,而长寿命的T₁态擅长远程能量转移,但二者难以协同优化。过去的研究多依赖材料固有性质被动实现单一途径,缺乏对双态功能的主动设计(图1a)。

新加坡国立大学江东林教授团队近日在《自然-材料》发表突破性成果,通过给体-受体(D-A)共价有机框架(COFs)实现了双态能量协同捕获。该材料采用卟啉为电子给体/三重态发生器,苯并噻二唑为电子受体(图1c),通过分离柱状π阵列、有序微孔和短D-A距离(1.2nm)结构(图2a),同步驱动S₁态电子转移与T₁态能量转移。独特的氢键网络(图1g)稳定激发态,使红光至近红外(1000nm)波段的光能高效转化为化学反应能,无需金属助催化剂或牺牲剂。
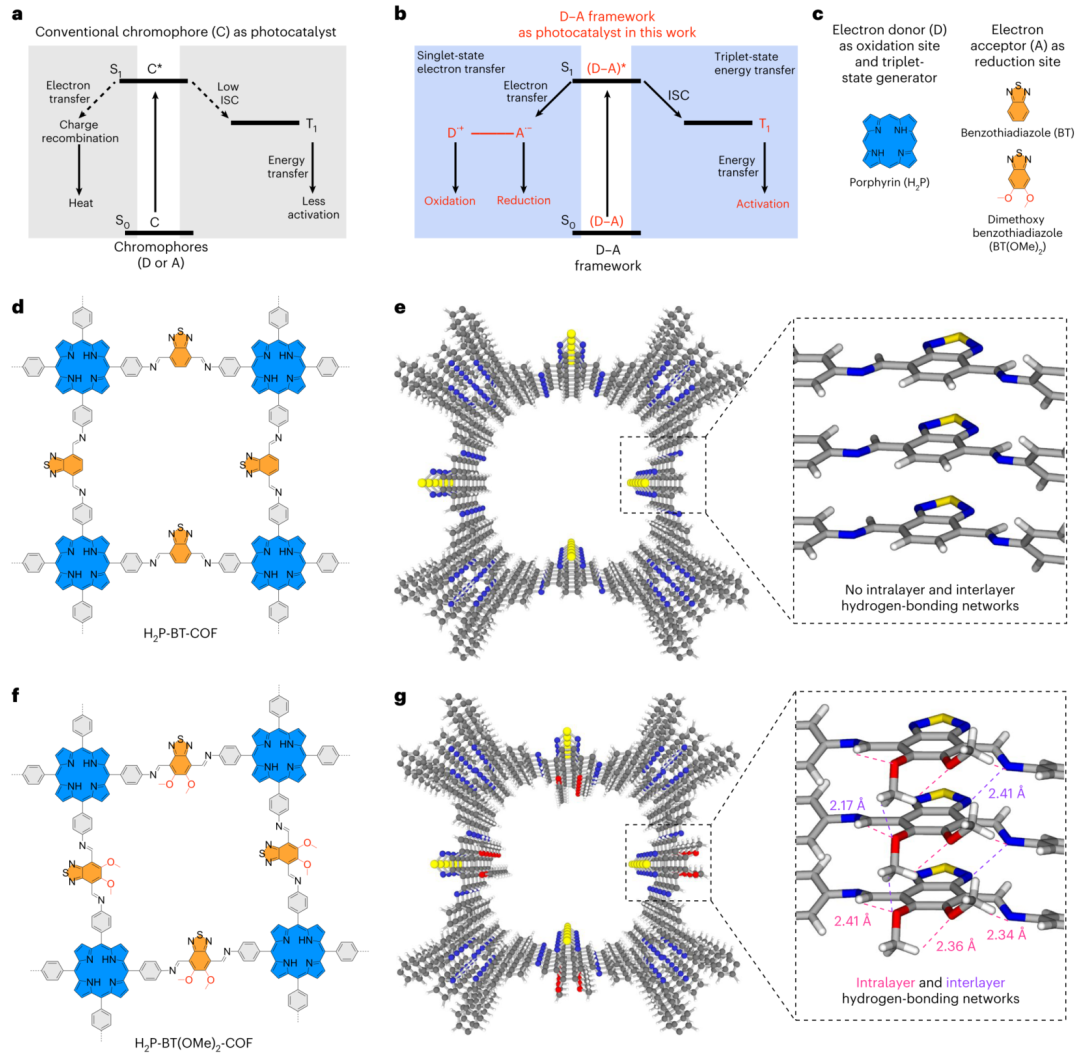
图1 | 光激发态与D-A框架光催化剂 a. 基于常规发色团光催化剂通过S₁或T₁态的光激发态与催化途径(星号表示激发态)。ISC:系间窜越。 b. D-A框架光催化剂中S₁与T₁双通道催化的光激发态。 c. 卟啉作为电子给体(D)、T₁态发生器及氧化位点,苯并噻二唑/二甲氧基苯并噻二唑作为受体(A)及还原位点的结构。 d,e. H₂P-BT-COF的化学结构(d)和重构晶体结构(e)(蓝色卟啉:给体;橙色苯并噻二唑:受体)。e图插图为无氢键存在区域。 f,g. H₂P-BT(OMe)₂-COF的化学结构(f)和重构晶体结构(g)。g图插图为发育完善的层内与层间氢键网络(紫色虚线:层间C-H···O/N氢键,2.17–2.41 Å;粉色虚线:层内C-H···O/N氢键,2.34–2.41 Å)。
材料设计实现双通道催化
研究团队合成的H₂P-BT(OMe)₂-COF(图1f,g)通过分子内/层间氢键(2.17-2.41Å)抑制分子振动,将S₁态寿命延长至6.67纳秒(图5)。粉末X射线衍射(图3a,d)证实其AA堆叠晶体结构,氮吸附测试显示568 m²/g的比表面积和1.73nm微孔(图3h,j)。高分辨电镜(图4e-h)直接观测到有序的卟啉给体柱(空穴传输)和苯并噻二唑受体柱(电子传输),一维孔道为反应物提供传输通道(图2b)。

图2 | D–A框架光催化剂的催化过程。a,H2P-BT(OMe)2-COF的重构单层结构。每个给体(D,蓝色)与四个受体(A,橙色)相连,每个受体沿x和y方向以1.2纳米的规则间隔与两个给体相连。天蓝色箭头表示S1状态下的光诱导电子转移,形成氧化和还原单元,这些单元作为氧化和还原中心,以促进光催化作用。较深的蓝色箭头表示从T1状态下的能量转移,以激活反应物并促进反应。Vis-IR,可见-红外;3O2,三重态氧。b. 重构堆叠层结构(五层):卟啉给体柱传输空穴,苯并噻二唑受体柱传输电子,有序一维孔道为反应物供给与产物释放提供通道(仅展示O₂与苄胺)。
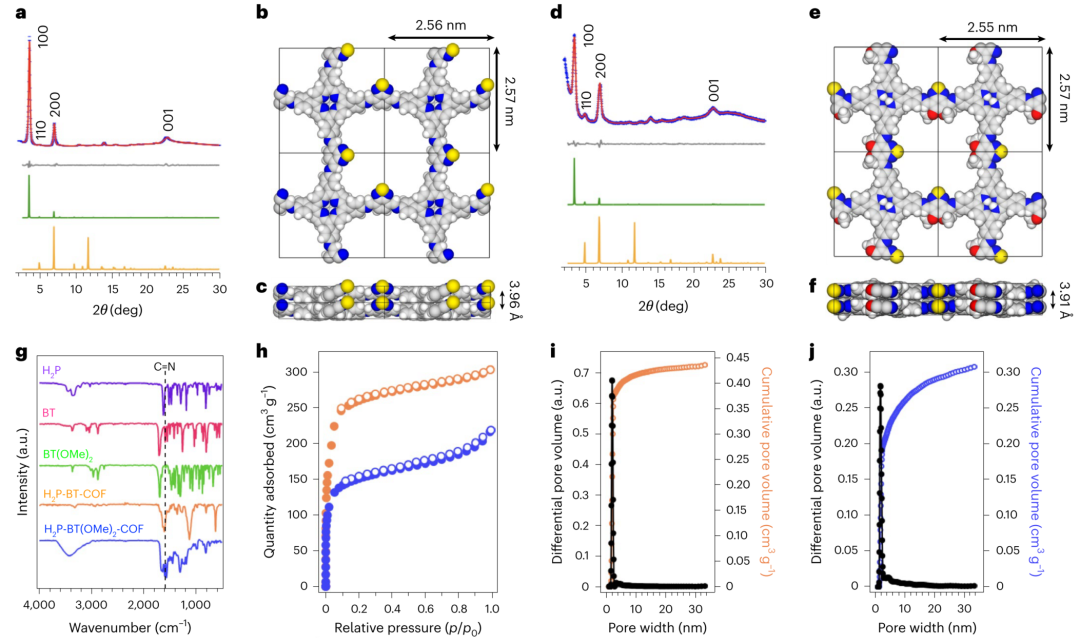
图3 | 晶体与多孔结构 a,d. H₂P-BT-COF(a)和H₂P-BT(OMe)₂-COF(d)的PXRD图谱(蓝十字:实验数据;红线:Pawley精修;灰线:差值;绿线:AA堆叠模拟;橙线:AB堆叠模拟)。 b,e. H₂P-BT-COF(b)和H₂P-BT(OMe)₂-COF(e)晶胞俯视图。 c,f. H₂P-BT-COF(c)和H₂P-BT(OMe)₂-COF(f)晶胞侧视图。 g. COFs与单体的FTIR光谱。 h. COFs的氮气吸附等温线(实心圆:吸附;空心圆:脱附;橙色:H₂P-BT-COF;蓝色:H₂P-BT(OMe)₂-COF)。 i,j. H₂P-BT-COF(i)和H₂P-BT(OMe)₂-COF(j)的累积孔体积(空心圆)与孔径分布(实心圆)。

图4 | 高分辨透射电镜(HR-TEM)分析 a–d. H₂P-BT-COF分析:HR-TEM图像(a,插图为FFT图像);IFFT图像(b);有序一维孔结构(c,俯视);堆叠层结构(d,侧视)。 e–h. H₂P-BT(OMe)₂-COF分析:HR-TEM图像(e,插图为FFT图像);IFFT图像(f);有序一维孔结构(g,俯视);堆叠层结构(h,侧视)。c,g图中红箭头指示分子单元位置。
光电性能超越现有体系
该材料光学带隙低至1.41eV(图5b),近红外吸收覆盖至1000nm(图5a)。甲氧基修饰将LUMO能级提升至-0.95V(图5c),增强超氧自由基(O₂•⁻)生成能力。电子顺磁共振(图5l,n)证实其红光照射下高效产生单线态氧(¹O₂)和O₂•⁻,DPBF降解实验显示140秒内完成96%转化(图5m)。载流子迁移测试表明材料具有双极性传输特性(电子/空穴迁移率比0.67),显著降低电子-空穴复合率。
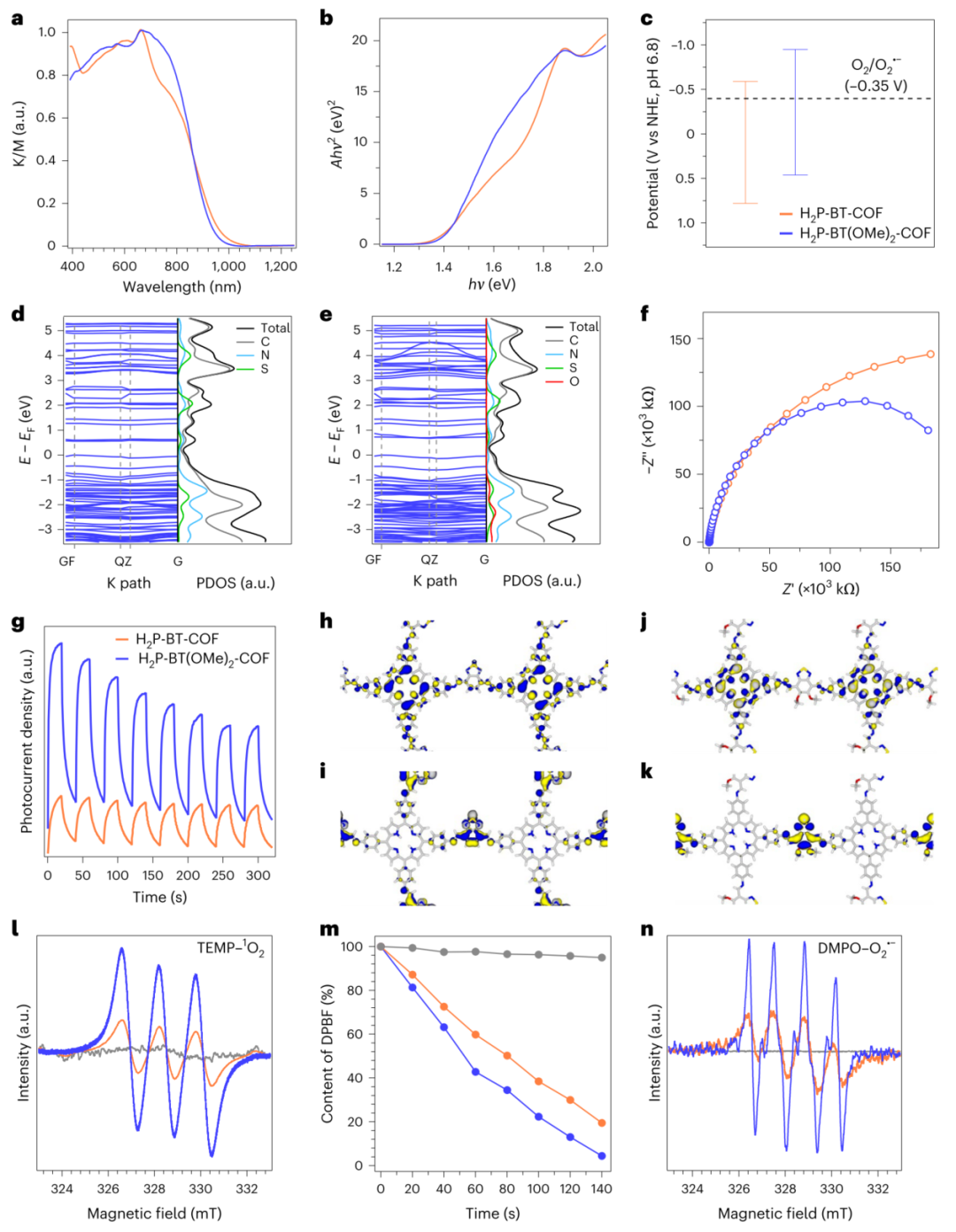
图5 | 光物理性质与反应中间体 a,b. H₂P-BT-COF(橙色)和H₂P-BT(OMe)₂-COF(蓝色)的固态电子吸收光谱(a)及对应Tauc图(b)。K/M:归一化Kubelka-Munk函数。 c. D-A COFs实验能级与氧还原电位对比(黑色虚线:-0.35 V)。 d,e. H₂P-BT-COF(d)和H₂P-BT(OMe)₂-COF(e)的能带结构(左)与投影态密度(PDOS,右)。 f. H₂P-BT-COF(橙色)和H₂P-BT(OMe)₂-COF(蓝色)的电化学阻抗谱。 g. 光电流密度随时间变化曲线。 h–k. H₂P-BT-COF(h,i)和H₂P-BT(OMe)₂-COF(j,k)的HOMO(h,j)与LUMO(i,k)轨道(蓝/黄色区域:轨道波函数相位)。 l. COFs在暗态(灰色)与红光(λ=620 nm)照射3分钟(含TEMP)的EPR谱图。 m. DPBF(40 μM)在无光催化剂(灰色)和有H₂P-BT(OMe)₂-COF(10 μg/ml,蓝色)/H₂P-BT-COF(10 μg/ml,橙色)条件下的降解曲线。 n. COFs在暗态(灰色)与红光照射3分钟(含DMPO)的EPR谱图。
催化性能刷新行业纪录
在苯胺氧化反应中(图6a),H₂P-BT(OMe)₂-COF的转化率达98%(>99%选择性),周转频率(TOF)高达1280 h⁻¹,比当前最优COF催化剂高3倍,比金属有机框架高10倍。其表观量子效率达31.6%(620nm),甚至在1000nm红外光下仍保持4.9%转化率。对于苯并咪唑合成反应(图6b)和C(sp³)-H活化/交叉脱氢偶联(图6c),TOF分别达55 h⁻¹和36 h⁻¹,均显著优于传统催化剂(如铱催化剂需10小时达到90%产率)。材料循环使用后活性无衰减。
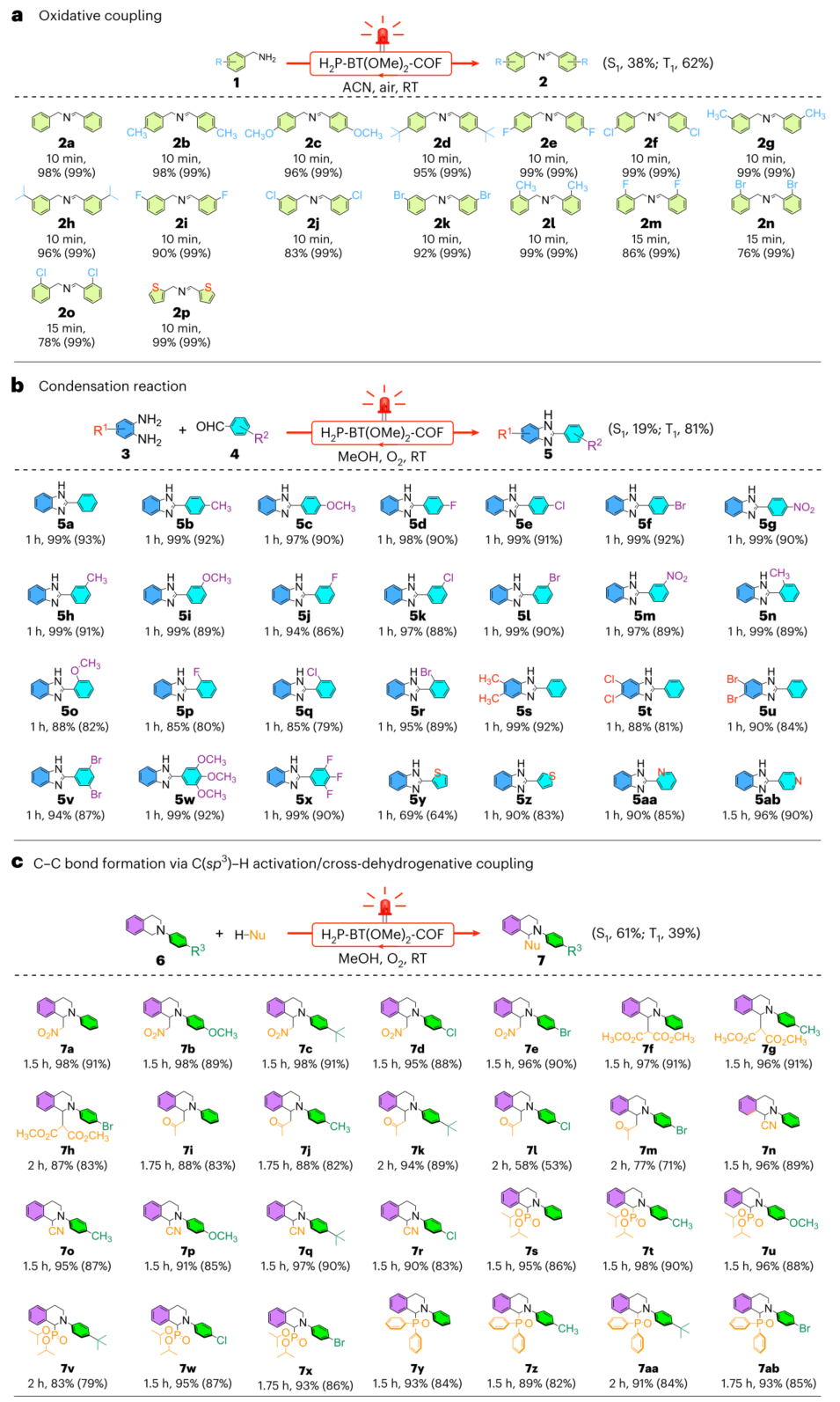
图6 | 光催化H₂P-BT(OMe)₂-COF的偶联与缩合反应 a. 氧化偶联反应(转化率经GC-MS测定,括号内为选择性),620 nm LED红光照射。ACN:乙腈;RT:室温。 b. 缩合反应(转化率经¹H NMR测定,括号内为分离产率),620 nm LED照射。 c. 通过C(sp³)-H活化/交叉脱氢偶联的C-C键形成反应(转化率经¹H NMR测定,括号内为分离产率),620 nm LED照射。Nu:亲核试剂。a–c图中右侧百分比为S₁与T₁态对反应的贡献率。
机制解析与普适策略
原位红外光谱(图7a)捕获到1205 cm⁻¹(内过氧化物)、1177 cm⁻¹(O₂•⁻)和943 cm⁻¹(¹O₂)特征峰,证实双反应路径并存。计算表明:电子转移途径中,甲氧基苯并噻二唑单元优先吸附O₂(吸附能-0.89eV);能量转移途径中,卟啉β位点主导¹O₂生成(吸附能-1.36eV)。不同反应中S₁/T₁贡献比例各异:氧化偶联反应为38%/62%,缩合反应为19%/81%,C-C偶联反应为61%/39%(图6)。

图7 | 光催化位点与反应机理 a. H₂P-BT(OMe)₂-COF在O₂氛围下的原位漫反射红外傅里叶变换光谱(DRIFTS)。 b,c. H₂P-BT(OMe)₂-COF上电子转移(b)与能量转移(c)至氧气的吉布斯自由能图。 d. 甲氧基加速双反应动力学示意图:极性甲氧基促进电子/能量转移途径(延伸数据图3)。
此项研究确立了将光激发态调控、能级优化与传质通道集成于一体的光催化材料设计范式,突破了太阳能驱动化学转化的固有局限。该策略为开发高效、无金属的广谱光催化系统开辟了新途径,有望推动清洁能源生产与环境修复技术的发展。
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。

官方微信
《腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606
- 邮箱:fsfhzy666@163.com
- 腐蚀与防护网官方QQ群:140808414

“海洋金属”——钛合金在舰船的

腐蚀与“海上丝绸之路”