
表面渗氮金相组织机理研究
2023-01-11 13:42:06
作者:每天学点热处理 来源:每天学点热处理
分享至:


钢铁零件在一定温度的可释放活性氮原子的介质中保温一定时间,使其表面渗入氮原子的过程叫作钢的渗氮或氮化。自1923 年,被德国人应用在钢铁上以来,渗氮工艺是目前对零件进行表面强化处理而广泛采用的技术之一。相对于常用的渗碳工艺,该工艺热处理温度低、畸变小,可明显提高零部件的表面硬度、耐磨性、那腐蚀性、抗疲劳性和抗咬合能力,有效延长了零部件的使用寿命。由于这些优点,渗氮处理广泛应用在汽车、船舶、航空等机械部件的生产中。
随着工业技术的发展,对工件表面强化的要求越来越高,渗氮工艺的种类也越来越多。但由于钢中含有不同程度的铝元素,形成的非金属夹杂具有过热敏感性,使渗氮层表面脆性增大,易发生剥落或开裂,同时,为了控制渗氮层的厚度以及渗氮层的致密性,对表面氮化层金相组织生长机理进行研究,以实现对不同工艺参数的调控,并弥补目前行业内对异常氮化组织生长机理缺少大量数据的现状,具有十分重要的意义。
通常以气体、熔盐或颗粒为氮化剂,工件表面渗氮或碳氮共渗一般采用480~590 ℃,硬度达到68~72 HRC。随着技术的发展,国内外开发出低温渗氮工艺,同时又为增加渗氮层厚度,提高渗氮效率,研究出表面纳米后的低温渗氮、循环渗氮、活性屏离子低温渗氮等。依据Fe-N相图(图1)[1],592 ℃时发生共析相变,γ-Fe(N)→α-Fe(N)+γ′-Fe(N)有奥氏体产生;650 ℃时发生共析相变,ε→α-Fe(N)+γ′ -Fe4N 在渗氮过程中,不同温度、不同氮浓度等参数下,会形成不同的相组织,各种不同相组织及化学式如表1所示。
表2 简单总结了渗氮过程及相变序列,渗氮层形成时,含氮铁素体(α-Fe(N))未达到饱和,随渗氮时间延长而增厚;随活性氮原子[N]原子的渗入和扩散,N 原子达到饱和发生相变,形成含氮奥氏体(γ-Fe(N)),以及奥氏体的相变,α-Fe(N)→γ-Fe(N)→γ′ -Fe4N;当γ′ 相达到过饱和状态时,形成含N 量更高的ε相(Fe2-3N)。最后渗层主要由γ′相和ε 相组成的化合物以及含氮铁素体组成的扩散层组成,在奥氏体渗氮过程中的渗层中还会存在由γ-Fe(N)组成的介于化合物层和扩散层之间的过渡层。碳氮共渗和渗氮的机理基本是相似的,但由于碳的加入,使表面形成的相更为复杂,目前对于Fe-N-C 三元相图,还没有明确的铁素体碳氮共渗和奥氏体碳氮共渗的温度划分点。
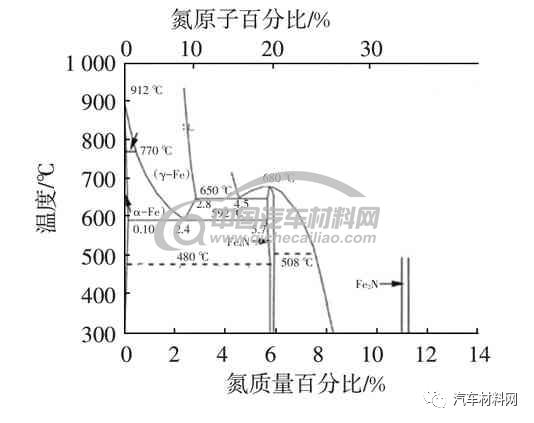
图1 Fe-N相图[1]
表1 Fe-N图中的相及性能
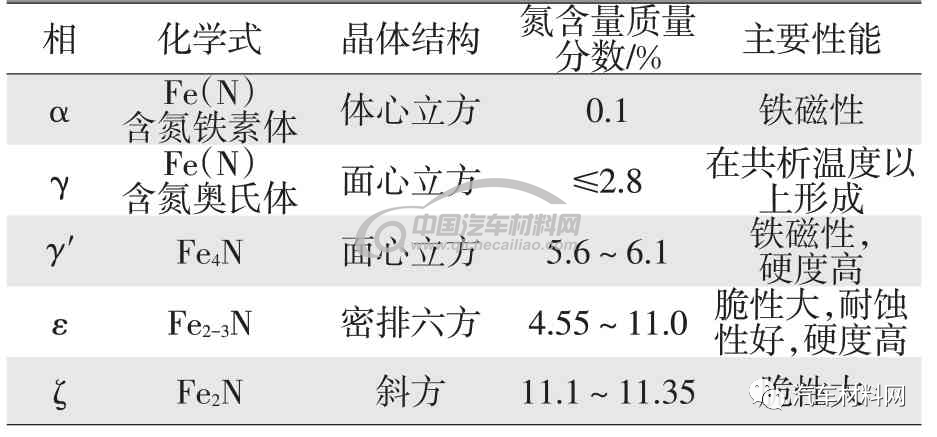
表2 渗氮相变序列及平衡态相组成

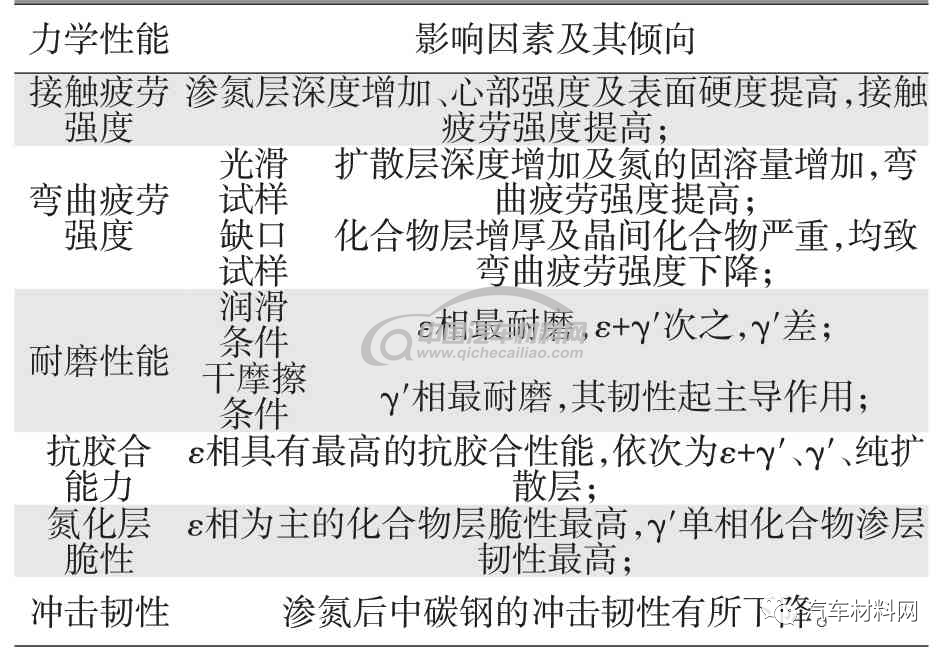
渗氮过程中不同的相是由不同的晶体结构为基体组成,不同的晶体取向会体现出不同的力学性能,如表3 所示,因此经不同工艺参数渗氮的零部件会体现出各种优缺性能。通过调控零部件表层的相结构,是实现提高零部件表面性能的重要手段,同时,通过研究各种相的组织形态特征,也是实现对组织的判定和创新工艺的重要途径。
表3 与力学性能相关的渗氮因素
3.1 γ′相金相组织特征
γ′相是以Fe4N 为基的固溶体,为面心立方的间隙化合物,在590 ℃左右,γ′相在渗氮层中为终极相或过渡相;渗氮降温过程中,γ′相即是γ相的共析相之一,也是过饱和α-Fe(N)的析出相;该相硬度高、韧性好,具有铁磁性,且该相经研究具有择优取向性,有利于提高耐磨性能,所以在氮化层金相组织检验分析中,该相是关注相。
低碳钢渗氮组织,表层组织均呈现为典型的柱状由表及里的“栾晶”定向生长方式,未发现六方截面,图2为渗氮层的光学显微镜(OM)图,图3为扫描电镜(SEM)图片。据有关资料研究分析,这种异常组织和温度过高,化合物分解转变成奥氏体有关。化合物层的柱状区域为过共析的γ′-Fe4N 和γ-Fe(N)奥氏体相。温度>590 ℃时,在α-Fe 基体上,γ和γ′相先后在表面沿氮原子扩散方向(箭头指向)形核并生长,成柱状晶粒,如图4 所示;温度<590 ℃时,则没有奥氏体相,γ相在冷却过程中发生分解为屈氏体,其形态体现了氮的沿晶渗入,如图5 所示。由表及里,这种异常组织的不同区域呈现的硬度为表面硬度为800 HV0.3,白亮层区域为320 HV0.3,黑带区域(屈氏体)360 HV0.3,扩散层160 HV0.3,基体组织区域145 HV0.3,硬度的波动如图6所示。

图2 低碳钢渗氮金相组织(OM图片)

图3 低碳钢金相组织(SEM图片)

图4 γ'相金相组织(SEM图片)
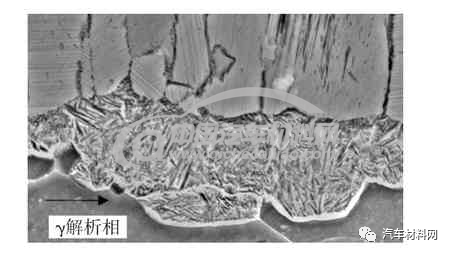
图5 γ'及γ相分解组织(SEM图片)
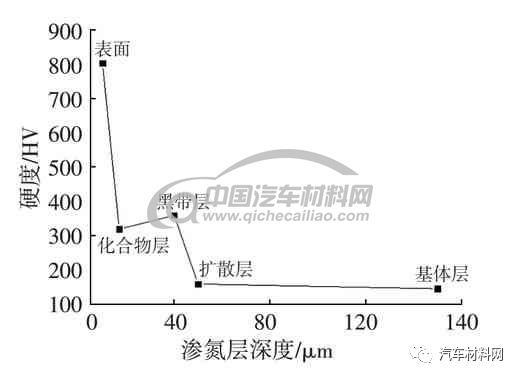
图6 硬度-层深
温度偏低和气氛浓度不足时会造成γ′相生长不充分。当温度<590 ℃时,图7 表层化合物未覆盖完全,最厚处约6.3 μm,并且γ′相层下没有γ相的共析类组织;图8 中黑色的片条状物质为马氏体加残余奥氏体,是奥氏体的过冷组织,由于氮气的气氛不足所造成,因为有奥氏体的分解产物,说明该工艺过程中温度>590 ℃。从浓度角度考虑,表层的间隙化合物为γ′相,不是ε相。
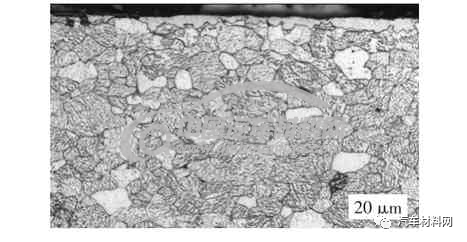
图7 γ′相不足产物(OM图片)
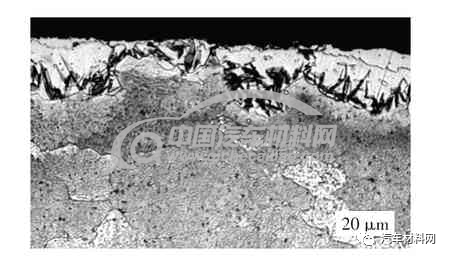
图8 γ′相不足产物(OM图片)
3.2 γ相组织及其分解相特征
图9及图10为低碳钢表面气体渗氮组织,白亮层和马氏体层的X射线能谱分析(EDS)及元素含量如图11、图12 所示。表面有极少量γ′相,白亮化合物层以γ相为主体,内层黑色物质为片状马氏体加残余奥氏体,该种片状马氏体为高氮马氏体,这种组织的形成是局部奥氏体化产生的,为奥氏体化后经过快速冷却的组织。表层残余奥氏体较多和马氏体未完全显示出来,与回火不充分有关,另外,奥氏体向马氏体的转变由于体积膨胀产生的内应力,也会抑制其转变。如在奥氏体冷变成马氏体的过程中,该种异常氮化组织形成的主要原是因为介质气氛浓度不足造成的。这种异常组织如内应力过大易于产生变形,使零部件发生开裂。

图9 γ分解金相组织(OM图片)
3.3 ε金相组织及其特征
ε相是以Fe2-3N 为基的固溶体,具有HCP(密排六方),氮原子浓度范围为4.55%~11.0%,所以在稳定的Fe-N 组织中,应该以ε相为主体,但是ε相为主的化合物层脆硬性较高,表层易剥落,为了提高其韧性,通常对表层进行调控。常规550 ℃渗氮表层的主要相为ε相和少量γ′相,图13、图14 金相组织及图15 X 射线衍射(XRD)谱显示,表层主要是ε-Fe2-3N 相和部分γ′相,图13 显示基体组织完全球化,主要是由于在氮化温度和氮化浓度较高,会引起组织奥氏体化的温度偏低,基体组织进入球化工艺状态。通常主要以ε相为主的白亮层组织形貌如图16 所示。

图10 γ分解金相组织(SEM图片)
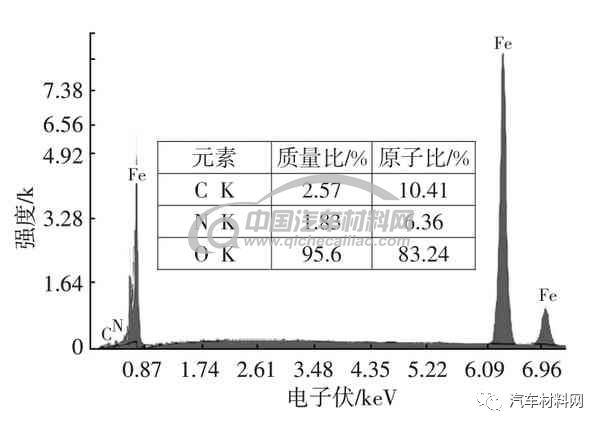
图11 白亮层EDS及元素含量
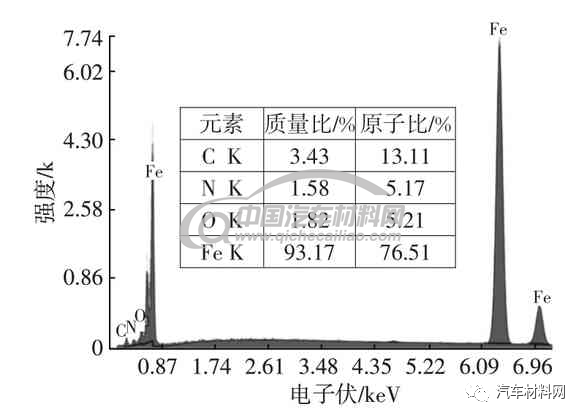
图12 马氏体层EDS及元素含量
3.4 α-Fe中的析出相组织特征
α 相为含氮铁素体,即氮在α-Fe 铁素体的间隙固溶体,590 ℃时,氮在α -Fe 中的最大溶解度为0.1%。渗氮过程也就是活性氮原子不断向基体中渗入和扩散的过程,当氮原子在α-Fe 铁素体中过饱和,以间隙化合物的形式析出γ′相,有很多不同的组织形态,如针状、颗粒状、脉状等。氮化物的析出位置一般为过共析钢,在晶界析出的比较多,在金相显微镜和低倍扫描电镜上显示为针状,如图17、图18 所示,但实际上该组织为类似于碳化物的条状或“刀片状”,主要是由于在某一特定晶界析出有关,如在高倍下的图19 及图20 所示;颗粒状、网状或脉状的析出氮化物不在晶界上,而在晶界内,各个氮化物类同于碳化物的析出,都是孤立存在的,如图19、图21~24 所示。

图13 氮化金相组织(OM图片)

图14 表层金相组织(OM图片)
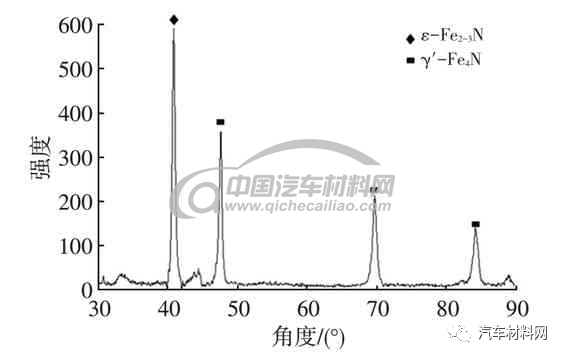
图15 表面XRD数据
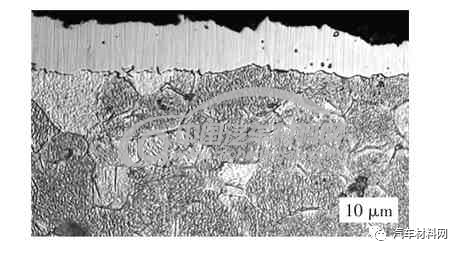
图16 ε相金相组织(OM图片)
α-Fe 中析出的各种相组织较为复杂,这与[N]的固溶量、扩散能力、析出条件等有关,但颗粒或网状析出物沿晶面成一定规律排布,如网状类似贝氏体组织的片层析出(图22),板条状的等距、有序排布,均显示出了晶体的择优取向特性。有关文献研究发现[2],在低温渗氮过程中,在氮比例较小时,渗氮层组织以γ′ -Fe4N 相为主,有明显的(200)γ′ 择优取向。

图17 α-Fe中析出相(OM图片)

图18 α-Fe中析出相(SEM图片)

图19 α-Fe中析出相(SEM图片)

图20 α-Fe中析出相(SEM图片)

图21 α-Fe中析出相(OM图片)

图22 α-Fe中析出相(OM图片)

图23 α-Fe中析出相(OM图片)

图24 α-Fe中析出相(OM图片)
通过大量数据研究分析了渗氮过程中的各种相的金相组织特征以及生长机理,这些资料可以作为渗氮工艺形成相组织的判定依据,并对其生长机理进行深入研究,弥补目前行业内,渗氮工艺过程中氮化物金相组织,尤其是异常氮化组织研究数据较少的状况。
a.表面渗氮金相组织,主要形成有3 类,间隙化合物(ε相、γ′相、ζ相等)、γ相的转变组织、α-Fe相中的析出组织(针状、颗粒状、脉状或网状等)。以γ′相组织为主体,分析氮化物组织的多样性和复杂性;ζ相氮浓度变化较小,不易在检验中确定。
b.γ相的转变组织多为氮化物中形成的异常组织,为含氮屈氏体、含氮马氏体加残余奥氏体、也有类似于贝氏体形状,多由氮化工艺的氮势不足所致,造成了表面化合物无法稳定、均匀析出;这种异常组织的发现也会为表面氮化零部件的失效提供检验依据。
c.α-Fe 相中的析出组织为扩散层中的氮化物,产生于冷却过程,形状较为复杂,某些形状的析出物,如脉状、板条状沿晶面的有序排布,体现了该组织的择优取向,可为织构调控方案设计提供参数。
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。
相关文章

官方微信
《腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606-806
- 邮箱:fsfhzy666@163.com
- 腐蚀与防护网官方QQ群:140808414
点击排行
PPT新闻

“海洋金属”——钛合金在舰船的
点击数:8240

腐蚀与“海上丝绸之路”
点击数:6568