
三峡大学李东升组《CEJ》:构筑新型、高效、耐用的助催化剂用于光催化产氢
2024-04-23 13:09:07
作者:材料科学与工程 来源:材料科学与工程
分享至:


第一作者:乔秀清;通讯作者:兰亚乾、李东升
通讯单位:华南师范大学、三峡大学
论文DOI:https://doi.org/10.1016/j.cej.2022.140791

全文速览
助催化剂辅助的光催化制氢可以明显的提高光催化活性,但材料的稳定性较差。本文提出通过调节助催化剂的组成和结构来构筑高效、稳定的光催化助剂,利用原位碳化策略准确合成了一种奇妙的助催化剂,即强锚定在导电碳基质中的MoO2/Mo2C-C纳米颗粒,并将其用作CdS光催化析氢的高效、稳健的助催化剂。令人惊喜的是,所获得的MoO2/Mo2C-C助催化剂非常稳定,在大气条件下储存两年也能保持原始活性。优化的MoO2/Mo2C-CdS-0.3(MMCC-0.3)光催化剂,在无需补充二次牺牲剂的条件下,产氢活性为18.43 mmol h−1 g−1,展示出连续90小时测试(在15天内进行)的超稳定性。实验结果和DFT计算都表明,独特的MoO2/Mo2C-C助催化剂不仅具有最佳的氢结合能(ΔG*)、降低的d带中心及析氢反应动力学势垒,导电碳桥、高质量界面和暴露出丰富的CdS活性位点,协同作用增强了可见光吸收,促进了电荷分离,保证了MoO2/Mo2C-C-CdS光催化剂的优异活性和稳定性。这项工作不仅为实际应用提供了一种高效、低成本的助催化剂,而且为设计用于光催化剂及其他用途的坚固助催化剂提供了宝贵的见解。
背景介绍
随着全球工业化进程,人类对清洁能源的需求越来越大,光催化分解水产氢可利用太阳能和水制备氢能,满足可持续发展的要求,是一种极具前景的策略。为进一步提升光催化剂产氢活性,助催化剂修饰策略引起了人们关注。发展高活性、高稳定性非贵金属助催化剂对促进光催化产业化进程极具研究意义。
具有特殊电子构型的钼基材料在光/电催化剂中越来越受到关注,其费米能级(Ef)位于Mo元素的4d轨道内,氧化钼(MoO2)在电导率(~6×103S m-1)和金属性质方面表现出无与伦比的优势。然而,MoO2多通过高温煅烧工艺制取,颗粒易于团聚、活性位点暴露困难,对催化性能不利。此外,MoO2与H中间物种的结合能力较差,导致产氢动力学缓慢,设计和探索具有良好分散性、丰富暴露活性位点和最佳电子结构的高效MoO2助催化剂,对于构建高效的助催化剂具有重要意义。
本文亮点
1.采用前驱体热解及原位碳化工艺获得了一种高效、超稳定的MoO2/Mo2-C助催化剂。
2.经过优化的MMCC光催化剂具有显著提高光催化的活性,超过90小时的稳定性。
3.该工作首次通过助催化剂组分和结构优化,实现了d带中心调控,提升助催化剂的活性及稳定性。
4.该助催化剂综合性能优于贵金属Pt,在实际应用中显示出良好的前景。
图文解析
通过苯胺离子C6H8N+和钼酸根离子Mo3O102-在碱性条件下的静电组装生成Mo3O10(C6H8N)2·2H2O前驱体,以前驱体为自牺牲模板通过改变热处理温度得到具有不同组分和结构的助催化剂。结合热重、XRD确定了助催化剂的生成过程为:

显然,通过MoO2的部分原位碳化可以获得锚定在碳基体上的MoO2/Mo2C-C杂化物(MoO2/Mo2C-C),这有望在组合物之间形成高质量的界面。然后,通过超声混合MoO2/Mo2C-C助催化剂和所制备的CdS-NRs,获得了MoO2/MO2-C-C-CdS(MMCC)光催化剂。
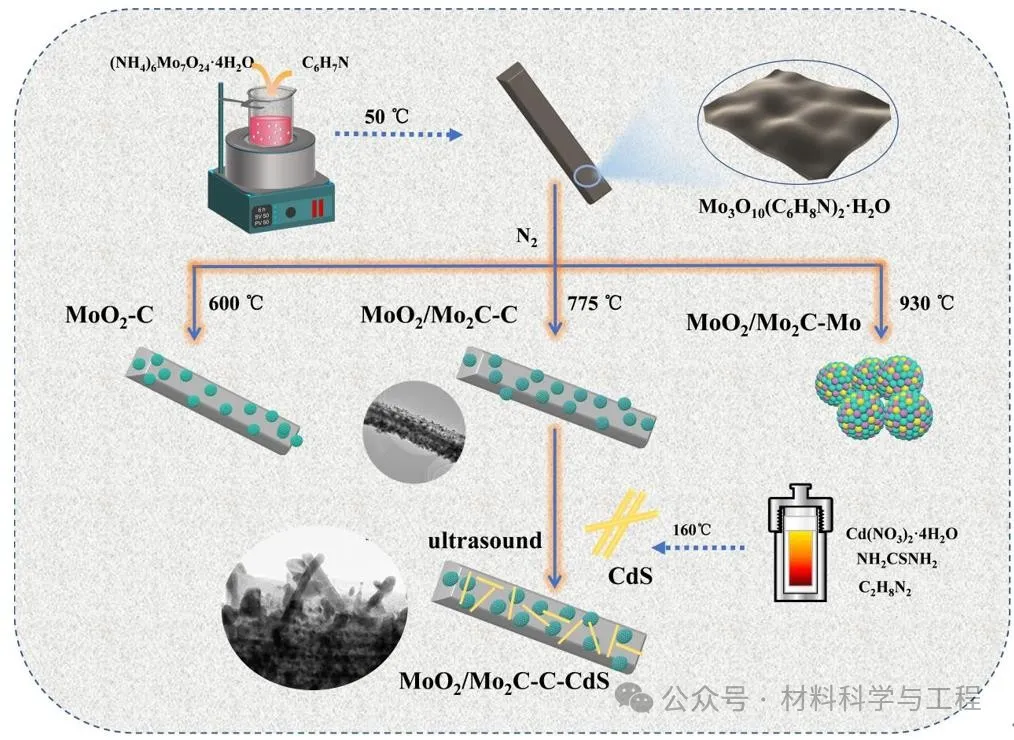
图1样品的制备过程示意图
XRD图谱和拉曼光谱证实了600°C、775°C和930°C下获得的产物分别为MoO2-C、MoO2/Mo2C-C和MoO2/Mo2C-Mo,单斜MoO2(JCPDS No.32-0671)和立方Mo2C(JCPDSs No.15-0457)及碳物种被观察到。SEM和TEM显示结晶、分散良好的MoO2/Mo2C-NP锚定在无定型的碳骨架上。图2g的间距为0.342nm和0.207nm的清晰晶格条纹分别归属于MoO2和Mo2C的(200)面。导电碳不仅防止助催化剂的聚集,还为电荷载流子的传输提供了“高速公路”,这将有利于电荷载流子的分离。
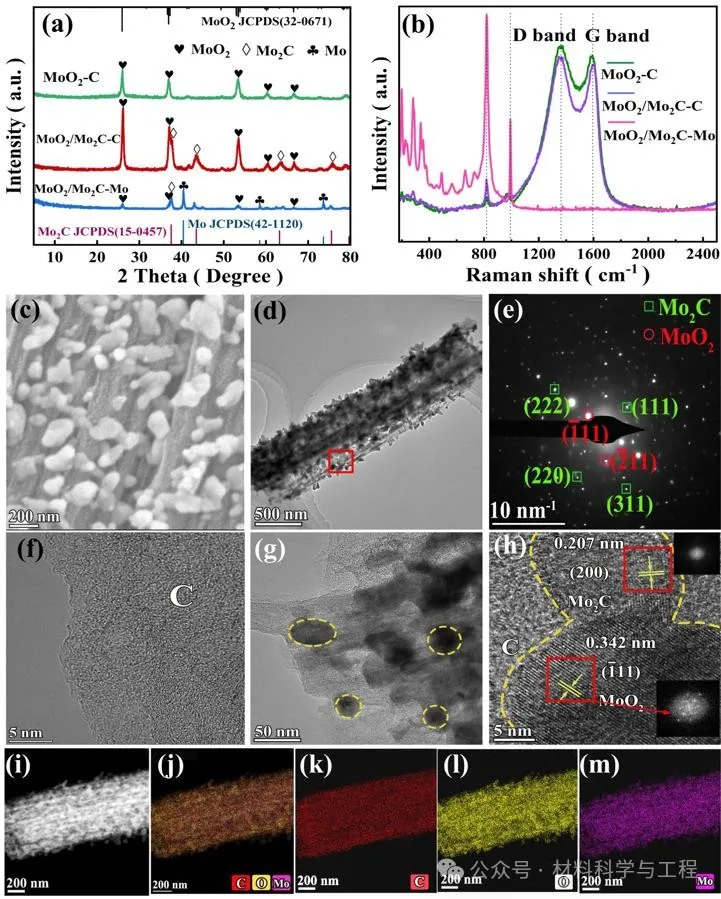
图2 MoO2/Mo2C-C的结构表征
应用Mo K边缘的X射线吸收结构(XAS)揭示了MoO2-C和MoO2/Mo2C中Mo物种的化学状态和局部配位环境。X射线近边缘吸收结构(XANES)表明,由于较高的氧化态,MoO2-C具有最高的能量吸收边缘(E0)。MoO2/Mo2C-C样品中Mo物种的E0位于Mo箔和MoO2C的E0之间,表明Mo带正电,平均化学价在Mo0和Mo4+之间。MoO2/Mo2C-C中Mo K边缘的总体特征与MoO2-C的特征非常相似,揭示了相似的配位结构。扩展XAFS(FT-EXAFS)曲线的相应傅立叶变换(FT)k3加权χ(k)函数表明,Mo箔在2.36Å处的峰值(未经相位校正)源于Mo-Mo反向散射配位。根据MoO2-C样品,在1.56和3.34Å处的两个主峰可分别归属于第一和第二配位壳中的Mo-O和Mo–Mo。2.17Å处的弱峰源于Mo-Mo的背散射。至于MoO2/Mo2C-C,1.56Å的峰可以与Mo-C/O的贡献相拟合,这很难区分,因为Mo–O和Mo–C的键长相似。然而,MoO2/Mo2C-C中较短的Mo–Mo键是由掺杂到MoO2中的C原子引起的。此外,与MoO2-C相比,由于Mo-C配位的散射贡献,该峰的振幅大大增加。2.14Å(Mo–Mo)和3.19Å(Mo-Mo)处的分辨良好的峰显示出与MoO2-C相似的散射路径。通过在R空间中对FT-EXAFS光谱的定量拟合,确定了Mo散射中心周围的C/O原子的配位数为2.00±0.34(图3c-d,表S1)。Mo-C/O壳层的平均距离为2.00±0.03Å。分析了样品Mo K边缘XANES的一阶导数,以揭示Mo物种的电子密度,图如图所示。第3e段。观察到的MoO2/Mo2C-C(20003.1 eV)的最大值位于Mo箔(20000.0 eV)和MoO2-C(20008.8 eV)之间,进一步表明Mo物种的氧化状态。图3f展示了Mo K边的半能和价态之间的关系。通过线性拟合,Mo在MoO2/Mo2C-C中的平均氧化态估计为+1.75,表明Mo处于缺电子状态。基于Morlet小波对Mo K边EXAFS振荡进行小波变换EXAFS(WT-EXAFS),以揭示R和K空间中的径向距离分辨率,如图3g所示,Mo箔在~8.2Å−1处的强度最大值可以归属于Mo-Mo的贡献。MoO2-C在~4.4和~12.3Å−11处的强度最大值归属于Mo-O和Mo-Mo的贡献。对于MoO2/Mo2C-C,在~6.5Å−1处的WT最大中心对应于Mo-C/O,而在~11Å−1的另一个最大中心与Mo-Mo贡献相关,证明了样品中MoO2和Mo2C的形成。
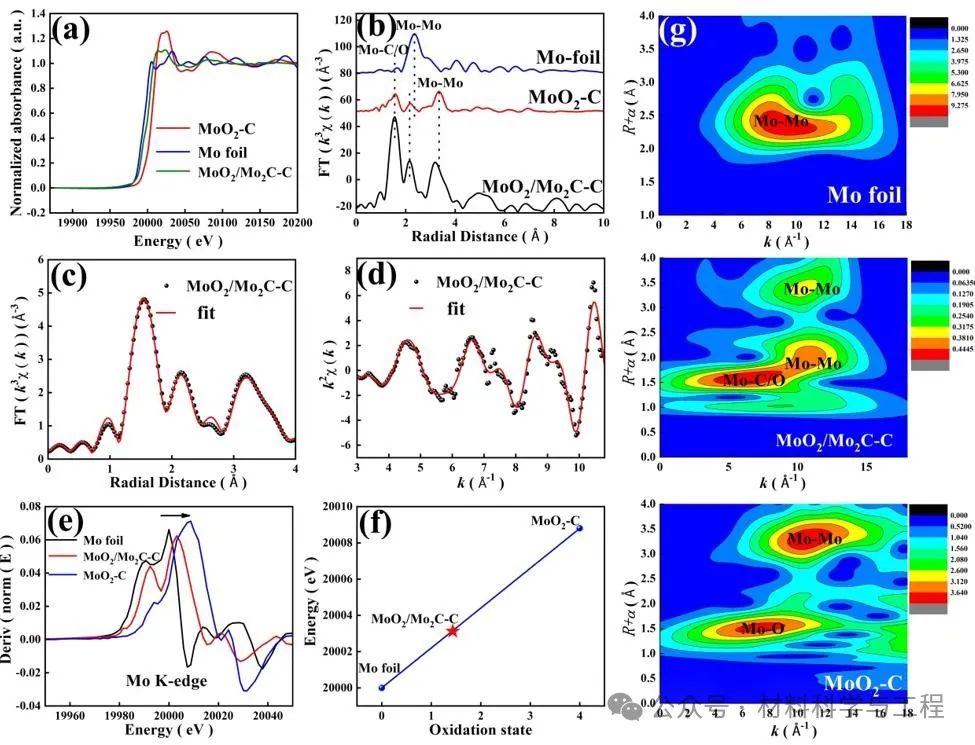
图3 MoO2/Mo2C-C的表征
为了评估MoO2/Mo2C-C助催化剂的活性,评估了MMCC样品的光催化析氢活性,并对MoO2-C、MoO2/Mo2C-Mo、C和商业铂碳改性的CdS进行了比较研究。单独的CdS表现出3.78mmol h-1g-1的低析氢速率,而MoO2/Mo2C-C助催化剂没有检测到析氢。当MoO2/Mo2-C与CdS结合时,析氢速率显著提高,表明MoO2/MO2-C仅作为光催化析氢的助催化剂。在MMCC光催化剂中,MMCC-0.3样品提供了18.43 mmol h-1 g-1的最高H2产生率,这比裸CdS提高了4.87倍。值得注意的是,MMCC-0.3表现出比其他助催化剂改性的CdS高得多的光催化活性。耐久性测试表明在没有补充二次牺牲剂的情况下,MMCC-0.3在连续90小时的长期耐久性测量(在15天内进行)中显示出相同的析氢活性,表明其具有优异的稳定性。令人兴奋的是,即使在大气条件下储存2年,MoO2/Mo2C-C助催化剂的活性也能很好地保持。相反,对照样品的析氢速率在18小时内严重恶化。因此,Mo2C不仅可以提高MoO2的助催化活性,而且具有超长期的稳定性,这对实际应用至关重要。
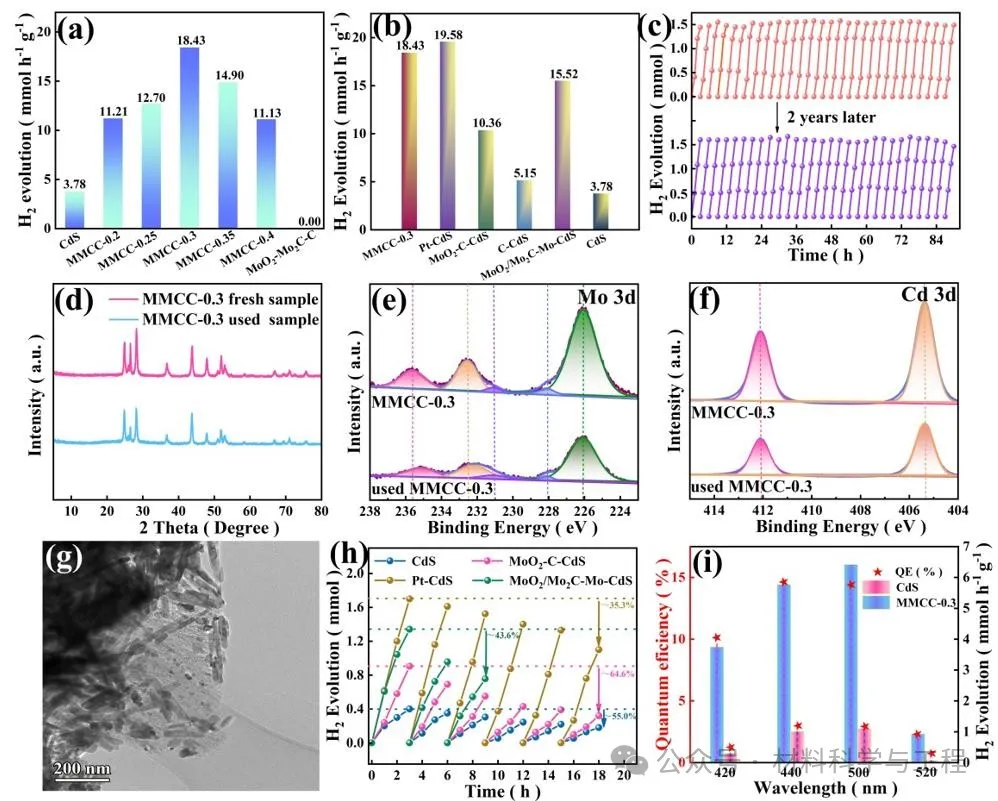
图4 MoO2/MO2-C-C-CdS(MMCC)光催化剂光催化产氢活性
界面处的差分电荷密度表明MoO2和Mo2C之间存在强烈的电子相互作用,电子从MoO2转移到Mo2C,从而优化了电子结构和|ΔGH*|,促进了光生电荷的分离。MoO2的费米能级穿过导带,验证了其金属特性。MoO2/Mo2C费米能级附近增强的局域态密度(图5g)表明了本征电导率的提高,从而保证了快速电子转移,有助于增强HER活性。MoO2/Mo2C异质结和MoO2的d带中心分别位于-1.27eV和-1.15eV,表明在Mo2C形成后,d带中心下移并远离费米能级。因此,反键态被减少,助催化剂和吸附质之间的相互作用减弱,促进了H*从助催化剂表面的解吸,从而促进了H2的析出。
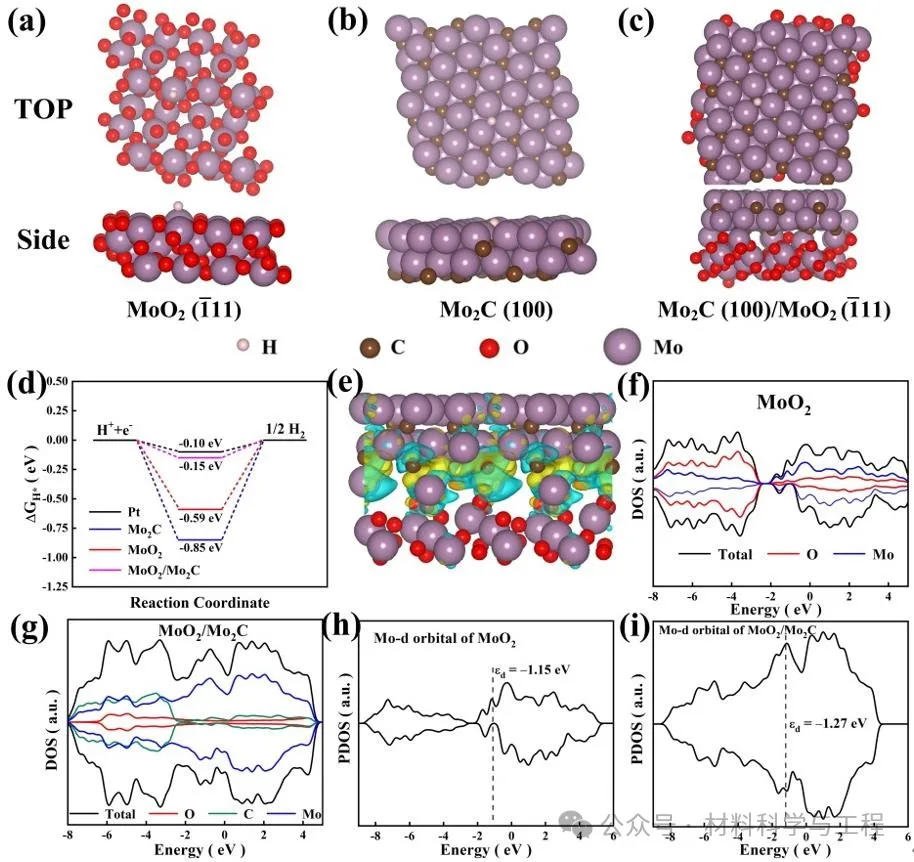
图5 DFT理论计算
样品的光电化学表征,证实了复合材料中增强的光吸收、电荷分离,这些因素共同作用提高光催化活性。
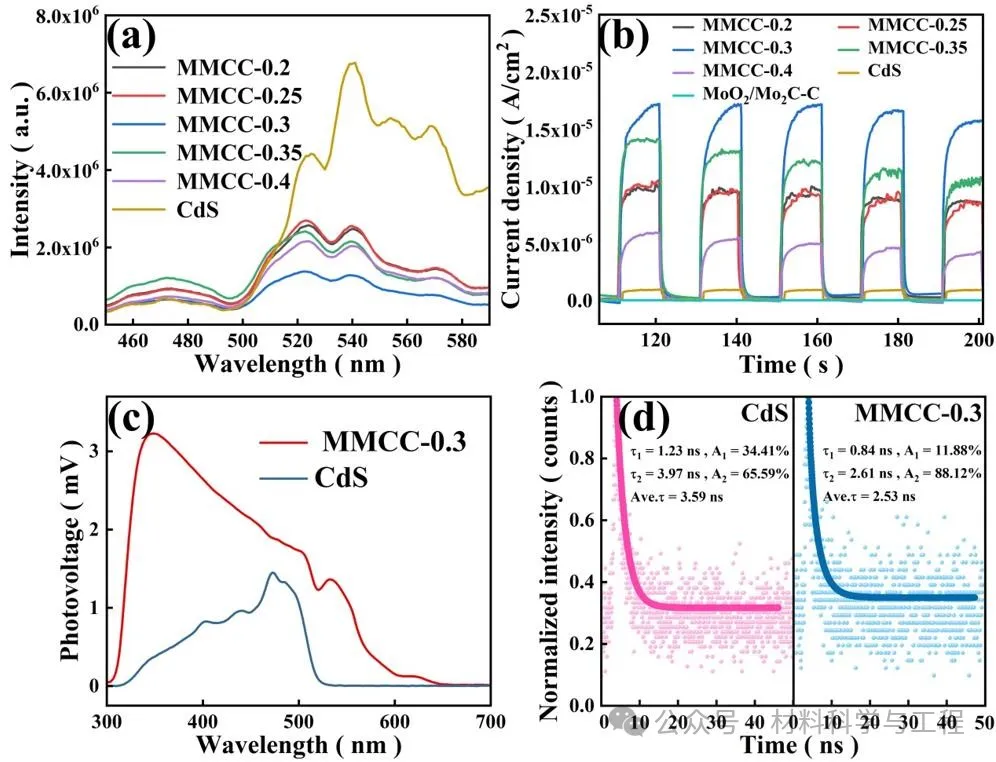
图6光电化学表征
全文小结
综上所述,作者成功构建了一种高活性、超稳定的MoO2/Mo2C-C助催化剂,用于CdS光催化析氢。杂化助催化剂中优化的氢结合能、d带中心和强电子耦合赋予了MoO2/Mo2-C优异的助催化活性。值得注意的是,MoO2/Mo2C-CdS光催化剂具有出色的HER性能和耐用性。证明了优化的助催化剂、高导电碳桥、各种组分之间的高质量界面、增强的光吸收能力和抑制电荷复合的协同作用,从而获得了优异的光催化性能。本文采用的策略为构建HER性能的其他有效助催化剂提供了有价值的见解。
通讯作者简介
兰亚乾:华南师范大学二级教授、博士生导师,教育部工程研究中心主任,英国皇家化学学会会士。中国化学会高级会员,中国化学会二氧化碳专业委员会副主任委员,中国化学会光化学专业委员会委员,中国感光学会光催化专业委员会副主任委员,中国化学快报(CCL)副主编,Natl. Sci. Rev.学科编辑组成员,Inorganic Chemistry、EnergyChem、Nano Research Energy、Polyoxometalates、物理化学学报、结构化学等期刊(顾问、青年)编委。主持国家自然科学基金杰出青年基金、优秀青年基金和面上项目等多项,近五年来以通讯作者在Nat. Synth.、Sci. Adv.、PNAS, Nat. Commun. (8)、J. Am. Chem. Soc. (13)、Angew. Chem. Int. Ed. (33)、Adv. Mater. (5)、Matter (2)、Chem (2)、Natl. Sci. Rev. (3)等期刊上发表通讯作者论文200余篇。论文被他引24000多次,ESI高引论文35篇,个人H-index 82。
李东升,博士/博士后,湖北省二级教授,博士生导师,教育部新世纪人才,享受国务院政府津贴专家、湖北省有突出贡献中青年专家;湖北省自然科学创新群体负责人,国家级学科创新引智基地负责人,中国复合材料学会矿物复合材料专业委员会委员兼副秘书长,三峡大学副校长;主要从事无机化学、能源化学、材料化学方面的研究,主持国家自然科学基金面上项目(6项)、教育部重点科研基金、湖北省自然科学基金创新群体等项目20余项,在Acc. Chem. Res.、Coord. Chem. Rev.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、ACS Catal.等期刊发表论文180余篇,被引14000余次,H因子59;授权国家发明专利56件,出版著作3部,获省部级科技一、二等奖6项; 入选爱思唯尔中国高被引学者和全球前2%顶尖科学家终身成就奖榜单。
第一作者简介
乔秀清:副教授,硕士生导师,浙江大学博士,主要从事光催化材料研究。主持国家自然科学基金1项,以第一作者在Chemical Engineering Journal,J. Mater.Chem. A, Chinese Journal of Catalysis等期刊发表SCI收录论文20余篇,论文被引1500余次,授权发明专利12项,指导研究生获得三峡大学优秀硕士学位论文。主持湖北省一流线上、线下混合式课程《材料科学基础》,指导学生多次获得“互联网+大学生创新创业大赛”湖北省金奖、银奖及铜奖等。
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。
相关文章

官方微信
《腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606
- 邮箱:fsfhzy666@163.com
- 腐蚀与防护网官方QQ群:140808414
点击排行
PPT新闻

“海洋金属”——钛合金在舰船的
点击数:9037

腐蚀与“海上丝绸之路”
点击数:7224